x

- info@bioicawtech.com
- Helsinki, Finland
- Request a quote
Sequencing analysis
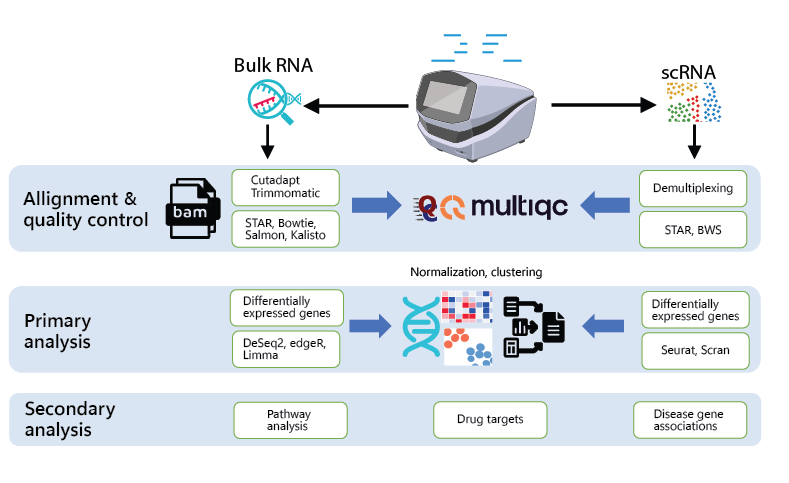
Scientists at BioICAWtech have developed several pipelines for analyzing bulk RNA, Whole exome sequencing and single cell sequencing.Following are some of the mostly commonly used steps by scientists at BioICAWtech.
- Raw Data Quality Control using FastQC
- Data cleaning using Trimmomatic and Cutadapt tools
- Alignment Tools
- Quantification
- Differential Expression Analysis (DE Analysis)
- Gene Set Enrichment Analysis (GSEA)
- Pathway analysis