Hamonizes and provide visual analytics on drug response data. Portal also integrates ML based live prediction of drug responses for uploaded any compounds.
Single-targeted drugs have demonstrated clinical success, it is unequivocal that single-targeted drugs often exhibit limited efficacy against complex diseases, such as cancers, whose development and treatment is dependent on several biological processes occurring simultaneously.
A comprehensive understanding of primary, secondary and inactive targets becomes essential in the quest for effective treatments for cancer and other indications.
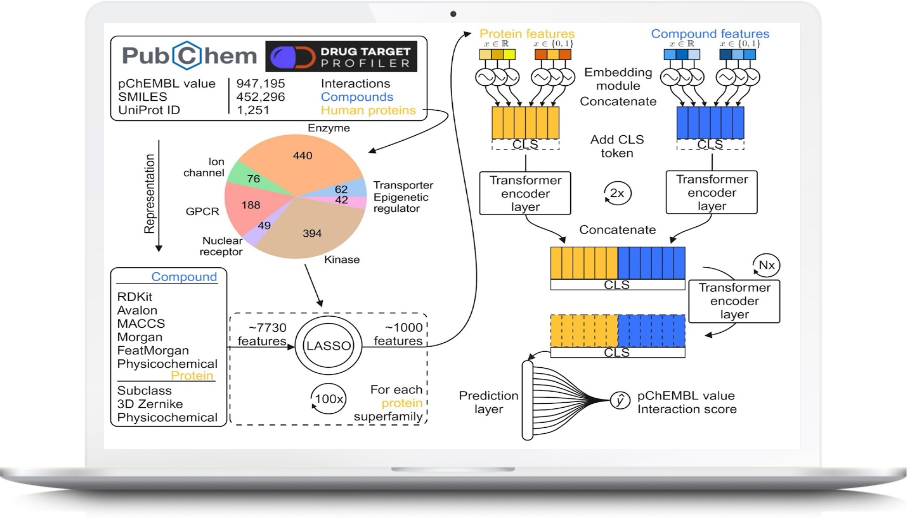
Figure: Workflow for developing attention-based models to predict bioactivities of approved drugs for targets across seven protein superfamilies. Active interactions are obtained from Drug Target Profiler, and inactive from PubChem. We generated a high-dimensional descriptor vector from the data to represent compound-protein pairs. We select the most relevant features with LASSO, resulting in one feature set for each protein superfamily. We repeat the selection 100 times for robustness and retain the most frequently chosen descriptors. We separate each set into protein and compound descriptors and pass them into specialized embedding modules. The embeddings are next concatenated, appended with a CLS token, and fed into a series of transformer encoder layers. Finally, the transformed CLS tokens are passed into a prediction layer to yield a pairwise binding affinity prediction. Each protein superfamily will have two models, one to predict pChEMBL values and the other to predict interaction scores (0-1).
Most FDA-approved drugs bind with only a small fraction of potent targets with average number of potent targets per approved drugs < 7 as reported in one of our scientist’s study (MICHA reference).
Therefore, our team members have recently developed AI based prediction method that is able to accurately identify any drugs/compounds for >1500 human protein targets across seven super families i.e. GPCRs, Kinase, Enzymes, Nuclear receptors, Ion channels, epigenetic regulators, and transporters. Nine different descriptor sets were meticulously examined to identify optimal signature descriptors for predicting novel drug-target binding affinities. Framework for our AI based prediction method across seven superfamilies is shown in the following figure.
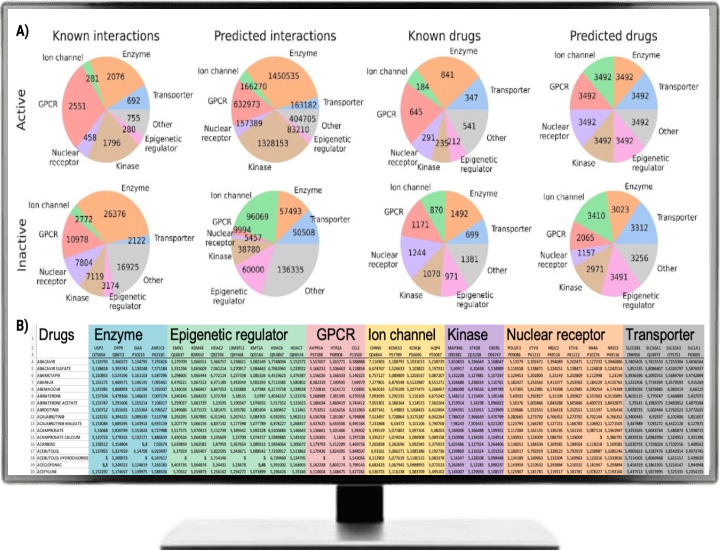
Figure: A) The number of known and predicted interactions and approved drugs for each protein superfamily. The proportions are separated into active and inactive categories. An interaction with a pChEMBL value of 5.0 is considered inactive. For the predicted values, we set the inactivity threshold at 5.1 due to the regression model predicting many values close to, but not exactly, 5.0. B) Complete drug-target matrix for known and predicted bioactivity values across seven protein superfamilies (1251 human targets in total)
Our proposed method has been rigorously tested on 0.2M endpoints from independent dataset and outperformed all state-of-art prediction methods for drug-target affinity prediction. Manuscript is currently review.
We are now further improving the method and extending the method to six additional superfamilies namely: Adhesion, Membrane receptors, secreted proteins, structural proteins, Transcription factors and Surface antigen. As per our information there is no such comprehensive drug-target prediction method that can systemically screen millions of preclinical compounds and approved drugs across 13 superfamilies of protein targets.
There are 3492 approved drugs (including salts) in ChEMBL33. But only a handful of approved drugs are experimentally tested against >50 human targets. Therefore, after successfully, testing and deploying drug-target based method, we went one step ahead and tried to complete drug-target profiles at full proteome (for 1251 human proteins). Figure 2A shows the pie charts for experimentally tested and predicted drug targets across seven superfamilies, whereas Figure 2B shows complete drug-target matrix for 3492 approved drugs (opening new avenues of drug repurposing).